

 In 2013, Ranbaxy USA Inc. pleaded guilty to felony charges and agreed to pay $500 million in fines for manufacturing adulterated pharmaceuticals at its Indian facilities.
In 2013, Ranbaxy USA Inc. pleaded guilty to felony charges and agreed to pay $500 million in fines for manufacturing adulterated pharmaceuticals at its Indian facilities.
Your product would be considered adulterated in the U.S. if you haven’t complied with good manufacturing practice (GMP) regulations in the manufacturing, processing, packing or storing of it.
Ranbaxy’s name is now infamous for its GMP violations. Sun Pharmaceuticals’ acquisition of the brand in 2014 effectively erased the Ranbaxy name. But Ranbaxy’s former owners were still facing lawsuits from their business partners five years later.
Ranbaxy’s case is just one example of how GMP violations can doom a company’s reputation. But the Ranbaxy case reached the U.S. court only after continued noncompliance, repeated inspections, warning letters and import alerts by the FDA.
The FDA escalates enforcement and consequences for GMP noncompliance based on the manufacturer’s response to a failed FDA inspection result. You can avoid most regulatory consequences by responding appropriately (related: GMP Compliance of Foreign Suppliers: The Importer’s Guide [eBook]).
Don’t let GMP noncompliance lead to serious disruptions and damage to your brand. Protect yourself by learning the consequences of failing an FDA inspection and the appropriate response to violations.
What happens after a routine FDA inspection?
At the close of a routine FDA inspection, an FDA inspector will issue an FDA Form-483.
An FDA Form-483, also known simply as a “483”, details any objectionable findings or violations discovered during the inspection. These violations are referred to as “inspection observations”.
The 483 doesn’t indicate the FDA’s official inspection result. But your supplier shouldn’t wait until the FDA’s official result to reply to the 483. Your supplier’s response to the 483 is their best opportunity to tell their side of the story and address how they will correct any violations before facing regulatory consequences.
The FDA will consider your 483 response together with their inspector’s Establishment Inspection Report (EIR) in determining further regulatory action.
The FDA will typically release an Establishment Inspection Report (EIR) within 30 days of visiting your supplier’s facility. This report will include the FDA’s official inspection result, categorized as one of the following:
- No action indicated (NAI) – No violations observed.
- Voluntary action indicated (VAI) – Inspectors found violations which don’t warrant additional regulatory action. Voluntary GMP compliance improvements are recommended.
- Official action indicated (OAI) – Inspectors found violations that require mandatory corrective action. The FDA will place regulatory and/or administrative sanctions on your business if these are not corrected.
The inspection is officially considered closed if inspectors issue your supplier an NAI or VAI result. And you can rest easy until the next routine inspection.
But if your supplier is issued an OAI result, there are mandatory steps they must take to become compliant and avoid further regulatory action.
Consequences you might face after failing an FDA inspection
You aren’t legally required to respond to a 483. But if you don’t, the FDA may take further regulatory action to prevent your business from distributing products in the U.S.
To avoid further action, any firm that receives a 483 should respond in writing to the FDA within 15 business days. In responding to the form 483, your supplier should:
- Address each observation noted in the 483
- Explain in detail how they plan to fix the issues
- Support their claims and responses with facts and data
 Quality and thoroughness of the response is extremely important. And clear documentation will help prove implemented corrective actions. As far as the FDA is concerned: “if you didn’t document it, it didn’t happen.”
Quality and thoroughness of the response is extremely important. And clear documentation will help prove implemented corrective actions. As far as the FDA is concerned: “if you didn’t document it, it didn’t happen.”
Your supplier’s response to the 483 directly impacts further regulatory action.
For some severe violations, the FDA might still take further regulatory action to protect consumers, even if the firm responds adequately to the 483. But in many cases, the form 483 is your best opportunity to clear your name and avoid further consequences.
How might your supply chain be affected if your supplier fails to respond adequately? Let’s take a look at the progression of the FDA’s enforcement actions:
1. Your supplier receives an FDA warning letter
A warning letter is the first escalation after the 483 and will include:
- A description of the violating conditions, practices or products
- The section of the law and regulation violated, where applicable
Anyone can view warning letters online at any time—including your customers. The initial warning letters and the company’s responses are publicly available on the FDA’s website, archived since 2012 with the name and location of the inspected facility.
The FDA will almost always issue a warning letter if your supplier has severe violations and fails to respond to the 483 in a timely manner. The FDA may also issue a warning letter if the supplier’s response to the 483 is inadequate or incomplete.
Crucially, the warning letter does not commit the FDA to any specific enforcement action. The FDA issues warning letters to prompt voluntary compliance from manufacturing companies—so you still have time at this stage to correct compliance violations before facing penalties.
Like the 483, the inspected facility must respond to the warning letter within 15 days to avoid further regulatory consequences.
How to respond to an FDA warning letter
After receiving a warning letter, work with your supplier to immediately draft a CAPA plan to address each violation listed in the warning letter.
 CAPA plans help your supplier:
CAPA plans help your supplier:
- Collect and analyze information
- Investigate and find the root cause of quality problems, specifically and systematically
- Take appropriate action to correct existing issues and prevent their recurrence
The warning letter will typically include suggested corrective actions for each violation. And if their earlier response to the 483 was inadequate, the warning letter will specify why.
Consider this 2018 warning letter for a drug manufacturer in Hangzhou, China:
In your response, you provided an updated standard operating procedure and the associated training records concerning the release of OTC drug products with inadequate quality oversight.
However, your response is inadequate because you have not determined the root cause of this violation.
The FDA also recommends the manufacturer conduct a root cause assessment and consider hiring a consultant to meet GMP compliance requirements.
Simply recalling a product or promising the FDA re-training to prevent future violations is often insufficient. Your supplier must show they understand how the problem occurred in the first place and provide evidence of corrective actions taken to fix it and prevent it from reoccurring.
Third-party auditing companies can conduct a follow-up audit after your supplier has had time to implement corrective actions to verify compliance. They typically can also help draft and monitor implementation of the CAPA plan and assess improvements.
2. The FDA places an import alert on your products
The FDA might place an import alert on your products if your supplier fails to adequately respond to a warning letter. The FDA might also issue an import alert prior to sending the warning letter if the violations pose a serious health risk to consumers.
 The FDA issues import alerts when the agency has enough evidence of violations to allow for Detention Without Physical Examination (DWPE) at the border. As of this writing, there are 88 active import alerts on products from China alone.
The FDA issues import alerts when the agency has enough evidence of violations to allow for Detention Without Physical Examination (DWPE) at the border. As of this writing, there are 88 active import alerts on products from China alone.
An import alert is issued to prevent harmful products from reaching customers in the U.S. Like warning letters, import alerts are also published on the FDA’s website.
Your product won’t be removed from an import alert until you take action to demonstrate GMP compliance. In fact, there are still active alerts from as early as 2011 listed on the FDA website.
For the FDA to remove an import alert for your product:
- You must submit a written petition to the FDA Division of Import Operations (DIO)
- The petition must provide evidence that objectionable conditions are resolved and give the FDA confidence that future entries will be compliant
- You must provide information proving a minimum of five violation-free entries shipped to the U.S.
- You’ll also need to submit an invoice, packing list and bill of landing for each entry you supply
Reviews can take up to 30 days to process before the FDA sends you a letter informing its decision.
3. The FDA requests or mandates a recall of your products
The FDA can ask a company to issue a recall if affected products have already reached the market. The FDA recalled 9,196 products in 2017, including:
- 3,609 food products
- 3,226 medical devices
- 1,176 drug products
FDA recalls are classified into three categories based on the risk posed by exposure to the affected products:
- Class I: Products likely to cause serious adverse health consequences or death
- Class II: Products that may cause temporary or medically reversible adverse health consequences but pose only a slight threat of a serious nature
- Class III: Products which aren’t likely to cause adverse health consequences but still violate FDA rules
Class II recalls were the most common type of recall issued in 2017 across all industries.
The FDA encourages voluntary recalls from importers in most cases. But under the Food Safety Modernization Act, the FDA can now issue a mandatory food recall. In these cases, the FDA must still give the responsible party an opportunity to conduct a voluntary recall before ordering a mandatory recall.
4. Courts issue an injunction (consent decree)
When it comes to GMP compliance, an injunction, or consent decree, is a legal order prohibiting a company from producing and distributing a product until violations are corrected. The FDA works with the Department of Justice (DOJ) to file a complaint in a Federal District Court and issue an injunction.
 Last October, the FDA announced a consent decree of permanent injunction against Keystone Laboratories, Inc. of Memphis, Tennessee for GMP violations. The injunction followed multiple 483s, an official warning letter in 2013 and re-inspections.
Last October, the FDA announced a consent decree of permanent injunction against Keystone Laboratories, Inc. of Memphis, Tennessee for GMP violations. The injunction followed multiple 483s, an official warning letter in 2013 and re-inspections.
Keystone routinely promised to take corrective actions, but the violations persisted. Now the company is prohibited from manufacturing or distributing any drug until the terms of the decree are met.
Companies subject to an injunction must meet the following conditions before resuming operations:
- Cease operations until corrective actions are implemented
- Hire a qualified independent expert to inspect their facility and ensure GMP compliance
- Establish and implement a comprehensive Quality Assurance and Quality Control (QA/QC) program
- Cover all costs associated with further FDA inspections and oversight
Injunctions are relatively rare—the FDA only issued 12 in FY2017. A consent decree is the last resort for the FDA to mandate compliance before banning a company from manufacturing permanently.
5. You could be criminally prosecuted
A company or individual that violates the Federal Food Drug and Cosmetics Act (FDCA) can face misdemeanor or felony charges.
Criminal prosecutions are increasing with the passage of the Food Safety and Modernization Act, which asserts companies can face consequences whether or not they intended to distribute adulterated products.
So even if you claim you were unaware of your supplier’s GMP violations, you could still be held accountable for selling and distributing adulterated products.
As with injunctions, the FDA partners with the DOJ to prosecute GMP violators. Criminal prosecutions can include:
- Felony charges under the Federal Food, Drug and Cosmetic Act
- Misdemeanor prosecutions of responsible corporate executives
- Prosecutions for fraud, money laundering and obstruction of justice
A single misdemeanor violation can equate to a $250,000 fine and a year in prison, while felony violations can lead to millions of dollars in fines.
6. The FDA could ban you from your industry
The FDA is legally required to debar an individual who has been convicted of a felony relating to the development, approval or regulation of any FDA-regulated product.
 A debarment is an enforcement proceeding that prohibits an individual or company from working in any capacity in an industry.
A debarment is an enforcement proceeding that prohibits an individual or company from working in any capacity in an industry.
Debarments specify a period of validity, with many being permanent. Others may last two, five, 25 years or somewhere in between, depending on the severity of the violation.
Individuals are typically subject to debarment, rather than companies. But the FDA debarred a company for the first time in March 2018.
Debarments are even rarer than injunctions—the FDA only in FY2018. Debarments usually occur only after a criminal investigation or when the accused individual has waived their right to a hearing.
How to ensure FDA inspection readiness at your supplier’s facilities
GMP regulations can be lengthy and hard to understand, even for native English speakers. Can you imagine how confusing the regulatory jargon likely is for your foreign supplier?
Here are a few tips to ensure FDA inspection readiness at your supplier’s facility and avoid severe disruptions to your supply chain:
Rely on a GMP audit checklist to outline requirements
Many importers rely on a GMP audit checklist to maintain GMP compliance and FDA inspection readiness at their supplier’s facility. A complete GMP audit checklist ensures factory staff understand your requirements and helps them prepare for future FDA inspections.
Most GMP checklists are organized by factory system and reflect requirements at various stages of a factory’s operations, such as:
- Organization and personnel
- Buildings and facilities
- Equipment and utensils
- Production and process controls
- Packaging and labeling
- Warehousing, storage and distribution
- Document and records controls
Creating a checklist helps prepare your supplier for an FDA inspection and helps you avoid the most common problems cited in a 483.
Conduct a pre-audit to assess GMP compliance
GMP pre-audits are a step-by-step assessment of your factory’s compliance with each requirement under GMP regulations.
Conducting a pre-audit can help familiarize your supplier with the FDA inspection process before an official audit. They’ll be better prepared to answer potential questions from the inspector and provide documentation to support their answers.
Importers often arrange a pre-audit of their overseas supplier through a third-party auditing company. The auditor will typically use their own internal GMP audit checklist based on the FDA’s requirements and current trends to assess compliance.
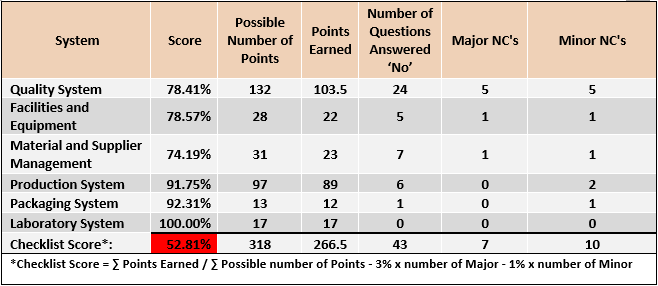
The audit report will clearly identify any observed violations broken down by each regulatory requirement, with the overall result typically categorized as:
- Compliant
- Needs improvement
- Not compliant
 Conclusion
Conclusion
As a buyer importing products into the U.S., it’s your responsibility to fully understand GMP regulations and ensure your suppliers are compliant. Many overseas suppliers won’t maintain FDA inspection readiness without some pressure from their buyers.
But a little due diligence and insight from a pre-audit can help you ensure your supply chain is FDA-ready before it’s too late.
Failing an FDA inspection can create a snowball effect of negative consequences for your company. Violations are often widely published and can damage your brand’s image.
Avoid the stress, damages and disruption that a failed FDA inspection can cause. Ensuring your suppliers are GMP-compliant may take a little extra work up front, but it’s sure to save you time and money in the long run.
How do you avoid the regulatory consequences of GMP violations in your supply chain? Share your experiences in the comment section below!




