

 In 2007, the former head of China’s Food and Drug Agency (CFDA), Zheng Xiaoyu, was executed for accepting bribes from manufacturing firms to register their sub-standard products without regulatory checks. At the time, a CFDA spokeswoman said, “As a developing country, China's food and drug supervision work began late and its foundations are weak”.
In 2007, the former head of China’s Food and Drug Agency (CFDA), Zheng Xiaoyu, was executed for accepting bribes from manufacturing firms to register their sub-standard products without regulatory checks. At the time, a CFDA spokeswoman said, “As a developing country, China's food and drug supervision work began late and its foundations are weak”.
With origins in the U.S. Federal Food, Drug and Cosmetic Act of 1938, good manufacturing practice (GMP) regulations in the U.S. and other Western countries have arisen from many decades of experience-driven efforts to regulate key industries.
But as the example above shows, China and other developing countries in Asia often don’t have this same strong foundation for holding manufacturers accountable for quality issues. At the same time, Western importers in GMP-regulated industries are increasingly outsourcing production abroad, as:
So what does this mean for you if you’re manufacturing a regulated product abroad?
Importers who manufacture their product abroad are still held accountable to GMP regulations. Companies that import substandard products into the U.S. can have the public alerted about their products, their products seized at the border and even face multi-million-dollar fines.
Your job as an importer is to educate your foreign suppliers on GMP guidelines and hold them accountable to your requirements. In this eBook we’ll explore why importers need to assess GMP compliance of foreign suppliers in the first place and how importers can evaluate suppliers to protect their business and consumers.
Read along, email a PDF to yourself for later by filling out the form on this page or click the links below to jump to the section that interests you most!
Good manufacturing practice (GMP) is a regulatory framework to ensure key products are consistently produced to quality standards appropriate for their intended use and product specifications.
Developed to protect consumers against unsafe and adulterated products, GMP regulations hold manufacturers in key industries like food, pharmaceuticals and medical devices legally accountable to quality issues in their products.
GMP regulations are a national framework, and national regulatory agencies are responsible for developing and enforcing GMP standards. These agencies include the U.S. Food and Drug Administration (FDA), Health Canada, the European Medicines Agency (EMA) and others.
Importers are only responsible for maintaining compliance with the GMP regulations for the target market they are intending to sell in. Each product type has its own regulatory document, which reflects the specific quality concerns and manufacturing processes for that product type.
 If you are intending to sell in many markets, U.S. FDA regulations are typically considered the “gold standard” for good manufacturing practice. U.S. compliance will typically give you access to most other markets. Thus, this guide will focus primarily on U.S. FDA requirements, but much of the advice is universal.
If you are intending to sell in many markets, U.S. FDA regulations are typically considered the “gold standard” for good manufacturing practice. U.S. compliance will typically give you access to most other markets. Thus, this guide will focus primarily on U.S. FDA requirements, but much of the advice is universal.
Regulatory agencies continue to amend GMP regulations periodically to reflect important changes in the regulatory climate. In the U.S., GMP standards are officially referred to as “cGMPs”, where the “c” refers to current. This is to impress upon manufacturers that they always need to compliant with current regulations and strive for continuous improvement.
Other countries simply use the “GMP” acronym, but they also require manufacturers to stay current with updated regulations.
Those importing the following products to the U.S. market must comply with FDA good manufacturing practices under Title 21 of the Code of Federal Regulations (CFR):
The FDA defines a medical device as any product “which is intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals.” This can include items like first aid kits, hospital beds and tongue depressors that you might not commonly consider “devices”.
Though not mandated, GMP compliance for cosmetic products is highly recommended in the U.S. And the FDA has the legal authority to inspect cosmetic facilities, as well as imported cosmetics.
Cosmetic GMP compliance is mandatory in the EU under European Regulation 1223/2009. Regulation 1223/2009 is harmonized with the ISO 22716 standard, meaning manufacturers can obtain ISO 22716 certification to prove compliance with EU legislation.
Experienced importers are likely familiar with ISO 9001 standards. Both GMP guidelines and ISO 9001 standards cover a factory’s quality management system (QMS).
But you shouldn’t regard GMP and ISO 9001 as equivalent standards, as they vary in their structure and purpose. Factories compliant with ISO 9001 requirements won’t automatically be compliant with GMP requirements, nor vice versa.
Let’s look at some of the key differences in the table below:
The focus of GMP is purely on product quality as it relates to product safety and efficacy. The focus is on protecting the consumer, or the patient in the case of medical devices and pharmaceuticals.
As you can see, ISO 9001 incorporates product quality into a manufacturer’s other important business needs. Meeting customer expectations and legal requirements is an important element of the ISO 9001 seven quality management principles. But ISO 9001 also focuses on operational leadership success, personnel engagement and continual improvement in addition to product quality goals.
It’s generally a good practice to ask your supplier for any local or international certifications they hold as part of the qualification process for new suppliers.
For GMP compliance, ISO 9001 certifications and other industry-specific certifications like ISO 13485 for medical devices and ISO 22716 for cosmetics are a good sign of a manufacturer’s GMP readiness. A factory that already has a well-developed, ISO-compliant QMS will likely find it easier to become GMP compliant than a factory with no experience in QMS standards. The factory will already have in place the structure, personnel and mindset for maintaining a QMS.
But you cannot use ISO certifications to verify GMP compliance, except for cosmetics in the EU. You’ll still need to cooperate with your suppliers to educate them on GMP guidelines and help them become compliant to pass a GMP audit.
Though ISO 9001 and GMP requirements differ in their mission and purpose, the goals of these frameworks can be complementary. If it’s important to your business that manufacturers comply with both ISO 9001 and GMP guidelines, be sure to educate your supplier on the differences in these two frameworks.
A quick online search of FDA import alerts and warning letters can reveal many GMP violations that should strike fear in importers’ hearts.
A June 2016 FDA inspection of a Chinese drug manufacturer in Yangzhou, Jiangsu province revealed several worrisome GMP violations:
 It’s the importer’s responsibility to pressure their foreign suppliers to care about GMP compliance, as well as educate them. The above supplier was probably unaware of the exact role of the FDA, as the FDA does not disclose details or so-called “trade secrets” gathered during inspections.
It’s the importer’s responsibility to pressure their foreign suppliers to care about GMP compliance, as well as educate them. The above supplier was probably unaware of the exact role of the FDA, as the FDA does not disclose details or so-called “trade secrets” gathered during inspections.
Think you’re not at risk if you’re only importing the product, not manufacturing it? Think again. In 2017, the FDA also issued a warning letter to Aztex Enterprises, a Canadian company that sourced drug materials from a supplier banned from importing to the U.S. The warning letter reads:
Because you used adulterated materials that you purchased from a firm on Import Alert … the drugs you manufacture also are adulterated under the FD&C Act.
You might be willing to risk that your supplier won’t be one of the facilities found to be noncompliant during an official FDA inspection. And you might think FDA auditors are less likely to inspect foreign facilities. But the importer actually bears the brunt of the consequences if their foreign suppliers aren’t compliant.
With the passage of the FDA Food Safety Modernization Act in 2011, food importers are now required to comply with the Foreign Supplier Verification Program (FSVP). Importers must verify that their suppliers meet applicable FDA food safety requirements, including that the food is not adulterated or misbranded with respect to allergens.
The FDA observed a failure to develop an FSVP in 108 food inspections in FY2017—its first year of mandated compliance. B&I Overseas Trading Company, a seafood importer, received a warning letter in April 2017 for a FSVP violation:
You do not have or have not implemented written verification procedures, product specifications and an affirmative step for ensuring that fish you import are processed in compliance.
Following FDA guidance, acceptable foreign supplier verification program activities may include:
Food importers should expect FSVP to be a continued point of attention in future FDA inspections. Importers should take steps to develop and implement an effective FSVP following FDA guidance documents.
Food and pharmaceutical inspections currently make up the bulk of foreign inspections conducted by the FDA. The FDA was mandated by the Food Safety Modernization Act to increase the number of foreign food facility inspections it conducts. Within one year of enactment, the law directed FDA to inspect at least 600 foreign facilities and to double those inspections every year for the next five years.
As more Chinese facilities register with the FDA, the FDA has begun to inspect more Chinese facilities. The number of drug manufacturing facilities in China increased 66 percent from 2011 to 2016.
In 2016, the FDA inspected 21 percent of 754 registered drug facilities in China. Objectionable conditions that warrant FDA regulatory and/or administrative sanctions were found at 22 percent of the inspected facilities.
The FDA recognizes that foreign inspections of medical devices are increasingly important, as imported medical devices now constitute 35 percent of the U.S. medical equipment market. U.S. Congress enacted the FDA Reauthorization Act of 2017 (FDARA) to reform the FDA’s inspection practices for medical devices and promote the adoption of a risk-based inspection approach.
The total number of foreign food facility inspections increased from 994 in 2011 to 1,363 in 2015. For drugs and devices, the number of foreign inspections has increased from 1,138 in FY2013 to 1,652 in FY2016. Beginning in 2015, the FDA conducted more foreign drug facility inspections than domestic. 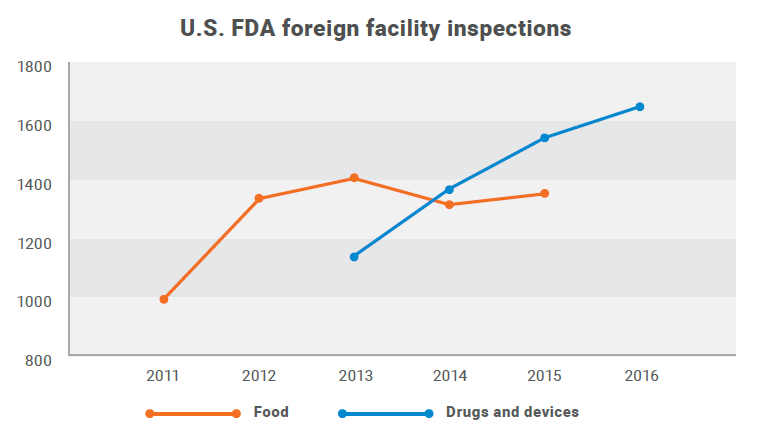
![]()
National regulatory bodies are increasingly working together to improve inspection efficiency and decrease inspection duplication. U.S. FDA and regulatory bodies in Canada, Australia, Brazil and Japan have joined the Medical Device Single Audit Program (MDSAP). After the successful completion of a two-year pilot program in 2016, the FDA now accepts MDSAP reports as a substitute for routine inspection FDA reports.
The EU has signed mutual recognition agreements with Australia, Canada, Israel, Japan, New Zealand, Switzerland and the United States covering most human medicines. The U.S. FDA’s only similar mutual recognition agreement is with the EU, signed in 2017 to cover most human medicines and active pharmaceutical ingredients (APIs). This means EU routine inspection reports can now be used to assess compliance with U.S. FDA requirements.
The FDA is not required to obtain the same level of documentation in foreign facilities as in domestic facilities to establish evidence of GMP violations during inspection.
The agency has the authority under the Food, Drug and Cosmetics Act to administratively restrict the importation of a product without demonstrating the adulteration of the product. Similar consequences apply if your supplier refuses to allow FDA inspections.
The FDA has the legal authority to enforce GMP guidelines through a number of mechanisms. These include:
Official FDA global enforcement statistics for fiscal year 2017 included: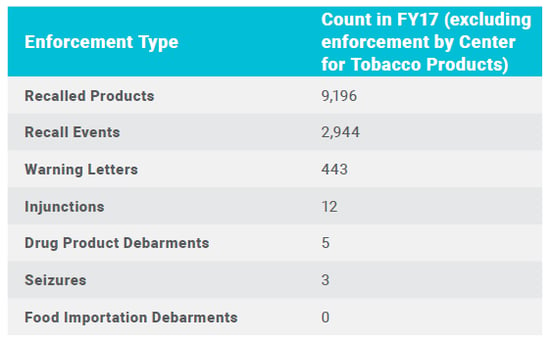
It’s harder to quantify the risk GMP violations pose to your brand’s reputation and customer loyalty. GMP regulations are in place for consumer protection, and the FDA publicizes all enforcement actions online. News that your product is unsafe or doesn’t perform as marketed can lead to negative product reviews, reduced sales and lower customer satisfaction and retention rates.
All foreign manufacturing facilities that manufacture products for import into the U.S. are required to register with the FDA and are subject to FDA inspections.
The FDA conducts four different types of inspections of manufacturing facilities, including:
The FDA chooses which foreign facilities to routinely inspect based on risk. This is to help ensure the FDA uses its resources effectively and efficiently to address the most significant public health risks.  These risk factors include:
These risk factors include:
Unlike domestic inspections, the FDA pre-announces foreign inspections. Factory management can notify relevant personnel and gather relevant documents ahead of the announced audit date.
Routine inspections are system-based, meaning inspectors will look at each system (records, equipment, personnel, etc.) individually and assess compliance. Foreign facility inspections typically last one to three days.
Depending on the FDA inspector’s preference, they might conduct an audit using one of three fact-finding methods:
If the inspector observes GMP violations on-site, the FDA will release an FDA Form-483, colloquially known simply as a “483”, within 90 days after an inspection. The 483 lists “inspection observations” or the violations found during the FDA inspection.
The overall result of the inspection is indicated as one of the following: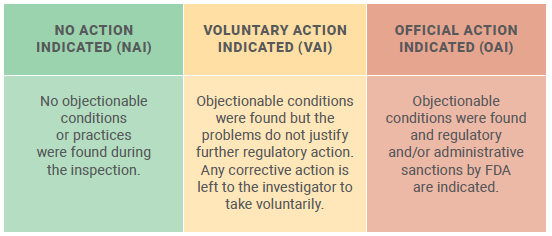
After issuing the 483, the FDA may issue a warning letter. A warning letter indicates that higher FDA officials have reviewed the inspection observations and that a serious violation at the facility may exist.
You can learn of some common GMP violations from official warning letters, import alerts and inspection observations published online by the FDA.
The FDA publishes a summary of inspectional observations for each fiscal year on their website. These are a composite of both foreign and domestic FDA inspection observations. Below are some of the common GMP violations observed in FY 2017 at food, medical devices and drug facilities:
As you can see, GMP violations often vary by product types. Sanitation issues are more commonly found in food facilities. Laboratory controls are only applicable to drug facilities.
Data integrity issues have been a recent focus for the FDA both domestically and abroad. The FDA defines data integrity as:
The completeness, consistency, and accuracy of data. Complete, consistent, and accurate data should be attributable, legible, contemporaneously recorded, original or a true copy, and accurate (ALCOA).
Data governance/data integrity deficiencies were cited in 67 percent of warning letters to drug firms outside the U.S. in fiscal year 2017. You can reasonably expect data integrity to be a focus in any upcoming FDA inspection, across all industries, both domestically and internationally.
For medical devices, inadequate procedures for corrective and preventive action (CAPA) was the most-often cited compliance issue in 2017. According to the FDA’s Inspection Guideline for CAPA, the purpose of a CAPA system is to:
Collect information, analyze information, identify and investigate product and quality problems, and take appropriate and effective corrective and/or preventive action to prevent their recurrence.
Many suppliers across all industries don’t implement CAPA well. Many suppliers fail to critically look at out-of-specification results to minimize their recurrence. And after an inspection has passed, many suppliers forget or otherwise neglect to follow up on CAPA. More focused on daily operations, manufacturers might feel they don’t have the time or resources to dedicate to CAPA development.
Factory staff can identify quality issues during manufacturing processes, during an audit or laboratory test or, in the case of customer complaints, after the product has hit the market. An effective CAPA system identifies and addresses all these quality issues with clear and documented steps to prevent their recurrence.
“It does not matter if it’s a Chinese or U.S. factory, most factories don’t have the discipline to implement, maintain and monitor a robust GMP compliance system,” says Kelvin Hooi, a GMP compliance professional with 20 years of manufacturing experience.
Your foreign suppliers are likely motivated by a desire to do business with Western customers but are not held accountable to your standards by local laws. As such, your supplier may see GMP requirements as a foreign imposition or requirement, not an ethical necessity. So taking proactive steps to assess GMP compliance of your foreign suppliers can be the only way to hold them accountable.
A pre-audit, often also referred to as a “mock audit”, is typically conducted by a third-party auditor to assess compliance to a standard, regulation or retailer framework. GMP pre-audits closely follow official GMP regulations to assess FDA inspection readiness.
GMP pre-audits can help importers:
Pre-audits are also a valuable opportunity to prep your supplier on the general process of a GMP audit before the official audit date. Relevant personnel can practice answering questions, gathering necessary documents and familiarizing themselves with the auditor’s processes.
For a first GMP audit, the exact audit path and procedures might not be available or known ahead of time. The FDA is not actually required to find all noncompliances at a supplier’s facility during a routine audit. Audit paths often vary based upon observations during the audit itself.
Always do your homework before a GMP audit. Evaluating the compliance history of your supplier can reveal systemic issues and provide an early warning of potential issues to focus on during the pre-audit.
A GMP audit checklist should be designed to identify all or most of what the FDA could identify during the audit. The official FDA audit path will vary from one facility to another and from one audit to another. Important factors to consider and integrate into a pre-audit include:
Based on trends in compliance issues gathered from other audits, the FDA might require auditors to include certain checks in all inspections. For example, if the FDA observes several facilities failing to monitor controlled temperature storage readings, then the FDA may require that inspectors include this check in all applicable inspections.
The compliance history of a facility will usually drive the audit process. FDA auditors will almost always follow up on previous observations to ensure issues have been adequately addressed. Importers and third-party auditors should monitor warning letters, Form 483s, import alerts and recalls from all previous FDA inspections at the supplier’s facility.
FDA auditors will check critical processes involved in the manufacturing of the product. Evaluate your supplier’s capabilities by finding out how much experience your supplier has in manufacturing this product for your market and what certifications they hold.
Along with compliance history, product complaints are a strong indicator of previous issues at the facility. As the importer, you should have a clear understanding of customer concerns. Evaluate product complaints to determine the origin of quality issues.
A pre-audit is typically conducted by one or two auditors with GMP experience over the course of two days. A pre-audit team should be aware of the current compliance climate for your industry and manufacturing country. These auditors should create a GMP audit checklist and plan the pre-audit path accordingly to focus on these factors and recognize associated issues.
A language barrier is a major hurdle in determining whether issues exist in records and reports, including data integrity issues. Naturally, if you’re importing from a Chinese manufacturer, all their internal documents are likely to be written in Chinese. Translation of these technical documents can be difficult (and remediation even more difficult) when it comes to identifying and addressing specific compliance issues.
If possible, work with a mock third-party auditor that has local and international expertise. Local expertise is important for identifying and translating supplier’s records, data, training plans and procedures. International or Western expertise is important to ensure the auditor is deeply aware of GMP requirements in your target market.
Ideally, you should audit potential suppliers before you begin working with them as part of the evaluation phase.
21 CFR Part 820.50 for medical devices states each manufacturer should:
Evaluate and select potential suppliers, contractors, and consultants on the basis of their ability to meet specified requirements, including quality requirements. The evaluation shall be documented.
After you’ve selected and started working with a supplier, it’s generally best practice to audit each supplier annually. According to the FDA’s Guide to Inspections of Foreign Medical Device Manufacturers:
It is recommended that the time between audits not exceed a 12-month period. Audits more frequently than 12 months may be needed if the firm has GMP problems.
If your first GMP audit finds the supplier to be compliant with few or no violations and the supplier has a history of GMP compliance, then annual auditing should be sufficient.
But you should determine the appropriate auditing schedule based on your specific concerns and situation. For instance, if a supplier is a sole source of an API with a complex manufacturing process, then experts suggest an assessment every six months.
Pre-audits are typically broken into step-by-step assessments of compliance with each requirement under an international standard. The findings for the factory’s compliance will often be categorized as:
Then the auditors will compile a final score of compliance from the number of each finding. Below is an excerpt from a pre-audit report for a medical device facility, showing a summary of all observations by requirement: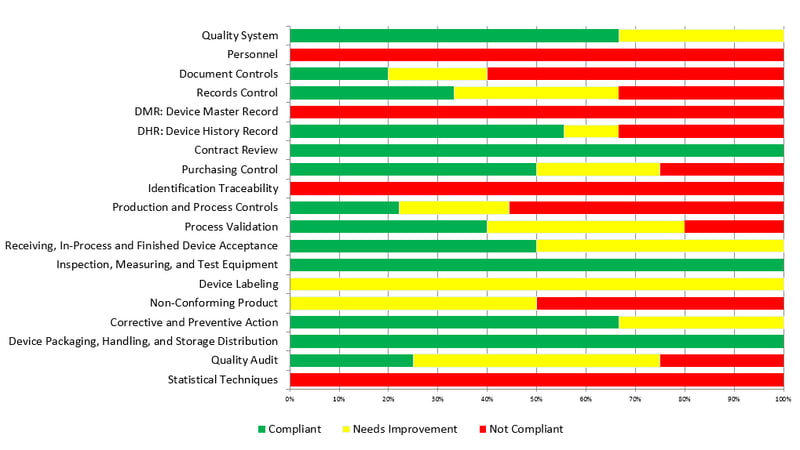
If a pre-audit reveals nonconformances, there are several steps you can take to ensure compliance before a potential official FDA inspection:
Not all suppliers who fail a GMP pre-audit are willfully noncompliant or uncooperative. In fact, many suppliers are eager to learn so they can better meet your requirements and increase their sales. In this way, GMP pre-audits can and do help foreign suppliers as much as they can help importers.
If you’re manufacturing abroad, chances are you don’t want to be traveling overseas every few months to monitor and maintain FDA inspection readiness. But you don’t want to fail an FDA inspection and face the consequences for noncompliance either. Now you know the consequences for importers with GMP violations are just as high as for domestic manufacturers.
With the help of pre-audits, you can identify issues at your suppliers’ facilities and proactively address them before an official GMP audit. Any supplier, whether in the United States, China, India or elsewhere, can pass a GMP audit and produce safe and high-quality products.
Ensuring GMP compliance at foreign facilities simply requires the time, effort and personnel to develop, implement and regularly audit a compliant quality management system.
Download InTouch's service guide below to find out how InTouch can help your importing business assess your foreign suppliers and become GMP compliant!





